{kind=link}

Deoxyribonucleic acid, or DNA for short, is the carrier of genetic information and is found in the nucleus of almost every cell in our body. We can imagine it as a “recipe book” in which individual recipes are represented by genes.
Thanks to genes, the cell can produce the resulting products, which are in most cases proteins. These subsequently perform various tasks in our organism, from construction, regulation, transport to many others. If there is an error in these recipes (genes), the resulting product may have different properties, or may not be formed at all. We refer to these errors as “mutations” and they are largely responsible for the various differences in our organism.
What exactly is written in these recipes was only discovered at the beginning of this century, when scientists read the entire human genome. It consists of approximately 3 billion building blocks, which we refer to as “nucleotides”. The entire DNA sequence is made up of 4 basic nucleotides – adenine, guanine, cytosine and thymine. For this reason, in the DNA sequence we encounter the letters A, G, C and T. However, the sequence of letters alone does not tell us anything. Today we know that genes make up only approximately 2% of the human genome and we have approximately 20,000 of them. The rest is more or less unknown, although we know that this part also contains important areas for the proper functioning of our cells.
Our DNA is 99.9% identical. At first glance, it may seem that we are almost the same. However, when we look back at the size of our genome, we find that we differ by about 3 million nucleotides. And it is precisely these places where different letters are found in the population that are of great interest to many studies. Thanks to these studies, we can now say, for example, that if you have a specific letter at a specific place in your DNA, then you have an increased genetic predisposition to developing a disease.
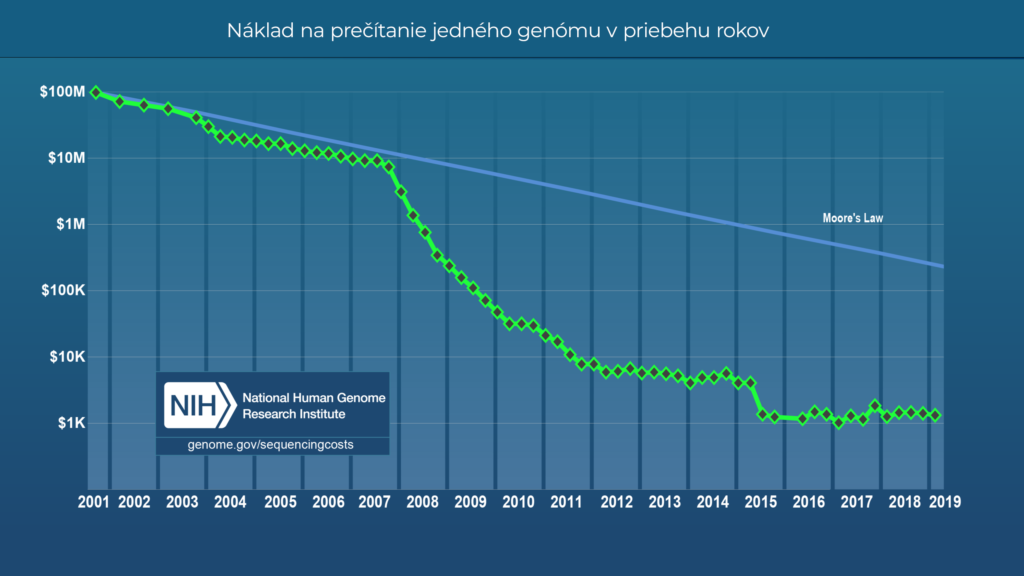
Thanks to the enormous progress in the field of “reading” DNA, almost everyone now has the opportunity to look directly into their cells, into the sequence of their DNA. This is truly a phenomenal achievement of science, since sequencing one human genome cost approximately 3 billion dollars at the beginning of this century, and it took 13 years. Today, we have already reached under 1000 dollars per genome, and the sequencing itself takes only a few minutes.
Currently, there are two main methods of reading DNA – sequencing and genotyping.
One of the techniques used to determine the sequence of DNA is so-called sequencing, i.e. determining the order of nucleotides in the genome of an individual. Currently, two approaches are mainly used, namely whole-exome and whole-genome sequencing.
Whole-exome sequencing technology focuses only on specific locations in the genome that provide information for the production of proteins. It is assumed that these parts, called “exons”, make up approximately 1% of the human genome. Together, all the exons in the genome are known as the “exome”, and therefore their sequencing method is known as whole-exome sequencing. This approach allows the identification of mutations in the coding region of any gene, and not just in a few selected genes. And since most known disease-causing mutations occur in exons, this sequencing method is effective in identifying such potential mutations.
However, it is known that mutations outside of exon regions can also affect gene activity and protein production, and thus can lead to various diseases. That is why the most ideal method is whole genome sequencing, which determines the order of almost all nucleotides in an individual’s genome.
Currently, the most commonly used method is the so-called ILLUMINA sequencing, which has generated more than 90% of all sequencing data in the world. The name of this method refers to the American company involved in the development, production and sale of systems for the analysis of genetic variations and biological functions, and which commercialized this method. This method is based on the detection of individual nucleotides (A, G, C and T) after they are incorporated into the growing DNA strand. Nucleotide detection is based on a fluorescent signal, meaning that each nucleotide is labeled with some kind of fluorescent label (we can imagine it as a colored marker) and after being added to the chain, the computer evaluates the given signal.

We are not yet at the point where it would be enough to read the entire human genome just once. Currently, sequencing methods are not yet accurate enough to be able to read the entire genome flawlessly in just one read. This is why ILLUMINA recommends a minimum coverage of 30x. This means that on average, each nucleotide is read independently 30 times.
However, some companies now also offer whole-genome sequencing with much lower coverage, in some cases even with only 1x coverage. With sequencing with such low coverage, it is currently unrealistic to read the entire genome. How is it possible to put the entire puzzle together when some pieces are not available from the read?
This process is quite complex, but in its simplicity it uses the principle of so-called “imputation”. In this way, unread sites are statistically filled in, based on known haplotypes in the population. This takes advantage of the fact that individual nucleotides are not inherited separately, but are inherited together in groups, which we refer to as “haplotypes”. However, in order to consider sequencing with such low coverage as relevant, it is necessary to have a large and population-specific reference panel, based on which it will be possible to fill in the missing parts of the individual’s genome with high accuracy. However, even when these conditions are met, this approach is not suitable for the analysis of very rare genetic variants.
In clinical practice, the entire genome or exome is often not sequenced, but only specifically selected sections – specific genes – are sequenced. This diagnostic examination is an effective tool for confirming or disproving a specific pathogenic mutation leading to a serious disease that is more common in the patient’s family history.
Despite the fact that whole-genome sequencing provides us with the best picture of our genome, it is still a relatively expensive method. That is why the so-called genotyping approach is currently used in commercial DNA tests, due to its effectiveness in terms of price/performance. It involves the analysis of specific sites in DNA on a chip. Unlike whole-genome sequencing, the entire DNA is not read, but only specific sites that were selected based on whether we know something about them and therefore whether we can associate them with something. These sites include, for example, mutations associated with various diseases or athletic potential.
As mentioned above, the sequence of nucleotides in the genome itself does not tell us anything; the key is the association of specific changes with specific traits.
Whole-genome sequencing provides the most comprehensive view of an individual’s genome, but it must be done in a high-quality manner, with sufficiently large coverage. However, this is still relatively expensive, and therefore, combined with the fact that we currently do not know the function of most sites in the genome, we cannot currently consider this approach effective for commercial genetic testing.
Genotyping does not analyze the entire genome, but only select sites with a known function are monitored, in which we often differ from each other. This effective approach has made it possible to reduce the price of genetic testing so much that it has become a suitable tool for commercial use. This is why we at DNA ERA also use this approach in our DNA tests.
Sources:
Photos: